Successful flow cytometry experiments begin long before a sample reaches the instrument. Proper sample preparation is one of the most important determinants of data quality, influencing signal resolution, background noise, and the ability to accurately identify cell populations. Even well-designed staining panels and optimized instrument settings cannot compensate for poor sample integrity.
The goal of sample preparation is to generate a clean, viable, single-cell suspension that accurately represents the biological population being studied. This requires careful handling, appropriate buffer conditions, and thoughtful experimental controls. By minimizing debris, preventing cell aggregation, and preserving antigen integrity, well-prepared samples enable the flow cytometer to measure cellular properties with high fidelity.
This technical note outlines the key principles of flow cytometry sample preparation, including strategies for maintaining cell viability, preventing clumping, optimizing staining conditions, and preparing samples for acquisition.
Fig. 1. Sample Preparation Workflow
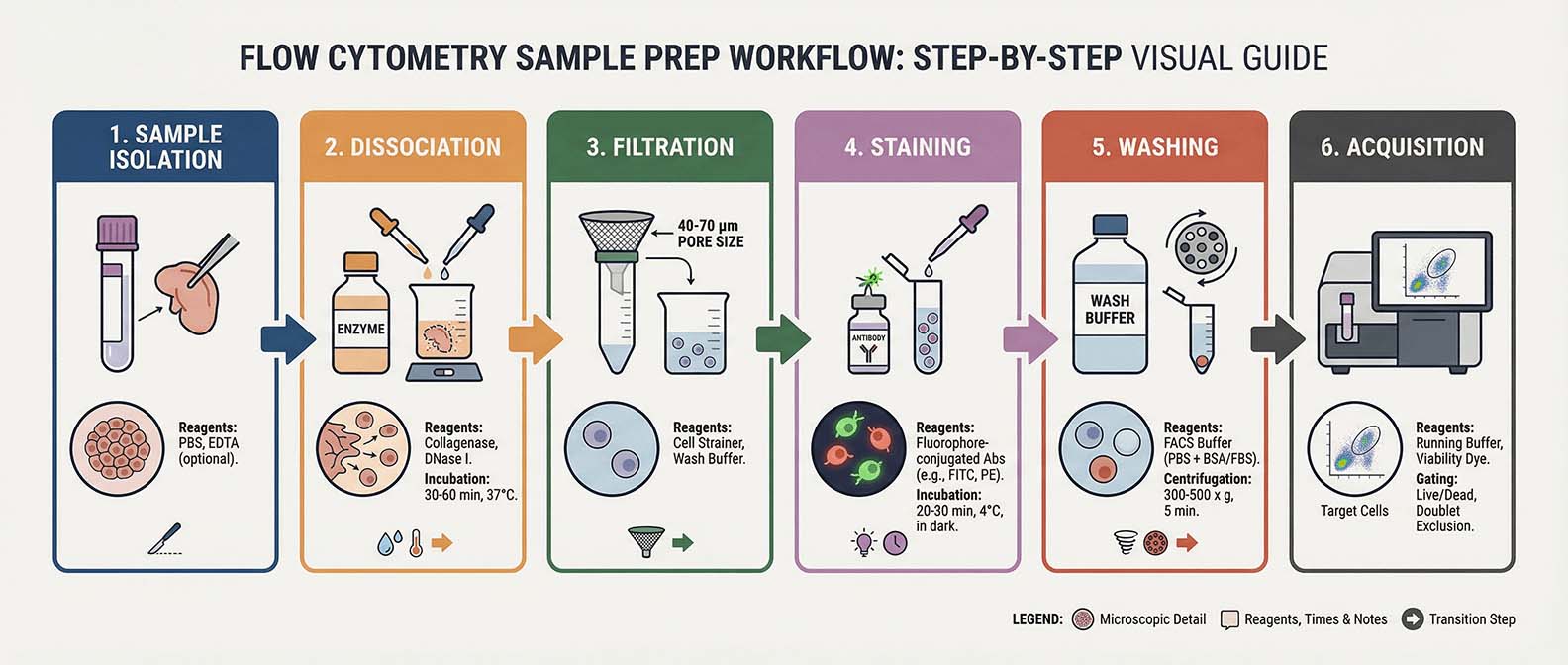
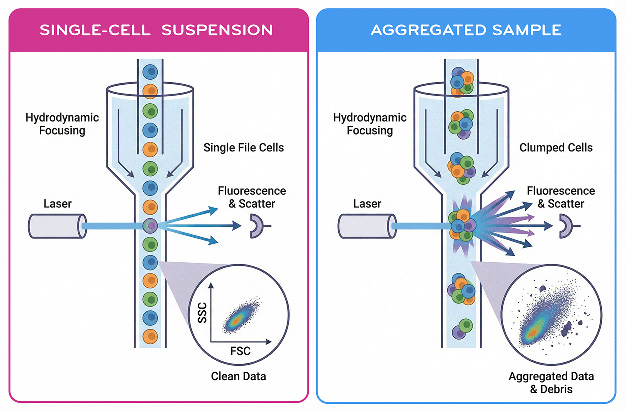
Flow cytometry measures cells individually as they pass through a narrow interrogation point. For accurate measurements, cells must therefore exist as separate particles in suspension. Aggregated cells can produce misleading signals because multiple cells may pass the laser simultaneously, creating artificially high scatter or fluorescence signals.
Achieving a single-cell suspension typically involves gentle mechanical or enzymatic dissociation, depending on the sample type. Suspension cultures often require minimal processing, whereas solid tissues must be carefully dissociated using mechanical disruption and enzymatic digestion. Regardless of the source, excessive shear stress should be avoided because it can damage cells and alter marker expression.
Filtering samples before acquisition is a simple but effective step for improving data quality. Passing the suspension through a 30–70 µm cell strainer removes clumps and large debris that may clog the instrument or generate inconsistent events. Gentle pipetting and periodic mixing also help maintain a uniform suspension.
The concentration of cells in the sample should be optimized as well. Overly concentrated samples increase the likelihood of coincident events, while extremely dilute samples reduce acquisition efficiency. Typical working concentrations range from approximately 1 × 10⁵ to 1 × 10⁷ cells per mL, depending on the instrument and experiment.
Fig. 2. Single-Cell Suspension vs Aggregated Sample
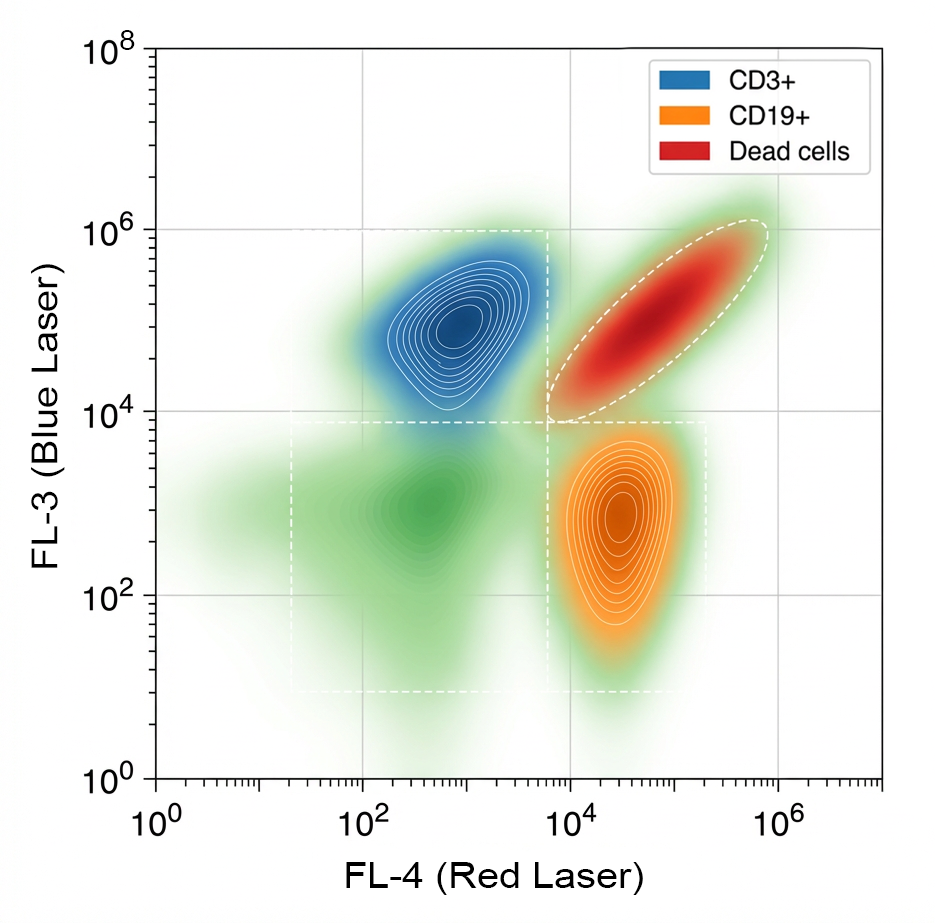
Cell health directly affects flow cytometry results. Dead or dying cells often exhibit altered scatter properties, compromised membranes, and increased nonspecific antibody binding. These artifacts can obscure true biological signals and complicate downstream analysis.
Maintaining cell viability begins with proper handling during isolation and staining. Cells should generally be kept on ice or at 4°C during preparation steps to slow metabolic activity and prevent internalization of surface markers. However, certain functional assays may require physiological temperatures to preserve cellular responses.
Appropriate buffer systems are also critical. Commonly used staining buffers contain phosphate-buffered saline (PBS) supplemented with protein such as bovine serum albumin (BSA) or fetal bovine serum (FBS). These additives reduce nonspecific binding and help stabilize cells during processing.
Incorporating a viability dye is strongly recommended in most experiments. These dyes discriminate live versus dead cells based on membrane integrity, allowing compromised cells to be excluded during analysis. Removing nonviable cells from the dataset significantly improves data clarity and reproducibility.
Fig. 3. Impact of Dead Cells on Data Quality
Debris and extracellular material can interfere with accurate flow cytometry measurements by generating background events or nonspecific fluorescence signals. Sources of debris include dead cells, tissue fragments, and damaged membranes released during sample processing.
Careful washing steps help remove soluble contaminants and excess staining reagents. Centrifugation speeds should be optimized to pellet cells without causing damage or excessive cell loss. In many cases, gentle spins of 300–500 × g for several minutes are sufficient.
Blocking strategies can also reduce nonspecific antibody binding. Cells that express Fc receptors, such as many immune cell types, may bind antibodies through Fc-mediated interactions rather than antigen recognition. Pre-incubating samples with Fc blocking reagents or serum can mitigate this effect.
Autofluorescence represents another potential source of background signal, particularly in activated cells or certain tissue types. While autofluorescence cannot be completely eliminated, careful fluorophore selection and appropriate unstained controls help distinguish true signal from background emission.
Accurate detection of cellular markers depends on efficient and specific antibody binding. Several parameters influence staining performance, including antibody concentration, incubation time, temperature, and buffer composition.
Antibody titration is one of the most effective ways to improve staining quality. Using excessive antibody can increase background fluorescence and reagent costs without improving signal resolution. Conversely, insufficient antibody may reduce sensitivity. Titration experiments identify the optimal concentration that provides maximal signal with minimal background.
Fig. 4. Antibody Titration Curve for APC Anti-CD3, clone OKT3
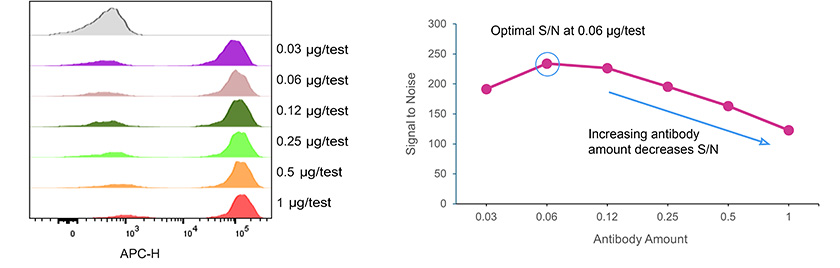
Incubation conditions also influence staining efficiency. Surface marker staining is commonly performed at 4°C for 20–30 minutes, which helps preserve antigen integrity and prevent receptor internalization. Following incubation, thorough washing removes unbound antibody and reduces background fluorescence.
For intracellular targets, additional steps such as fixation and permeabilization are required to allow antibodies to access intracellular compartments. These treatments must be carefully optimized because they can alter cellular morphology and fluorescence properties.
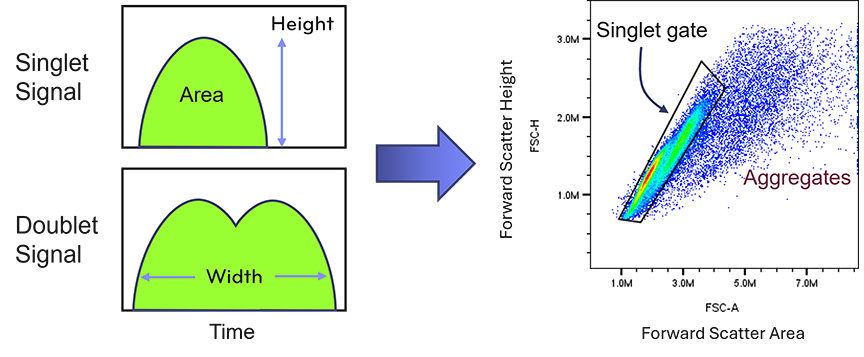
Cell aggregation is a frequent source of poor flow cytometry data. Aggregates can arise from DNA released by dead cells, sticky membrane proteins, or mechanical stress during preparation.
Inclusion of DNAse during tissue dissociation may help reduce clumping caused by extracellular DNA. Gentle resuspension and the use of appropriate buffers also help maintain single-cell suspensions.
Doublet discrimination during data analysis can remove some aggregated events, but preventing aggregation during preparation is the most effective strategy for preserving data quality.
Fig. 5. PBMC Doublets Versus Singlets
Before samples are run on the flow cytometer, several final checks help ensure reliable acquisition.
When large numbers of samples are analyzed, maintaining consistent preparation timing helps reduce variability across experimental conditions.
Reliable flow cytometry data depend on consistency across experiments. Standardized protocols, consistent buffer formulations, and careful documentation of preparation conditions help ensure reproducibility.
Key practices that support robust sample preparation include:
Attention to these details enables the flow cytometer to measure cellular properties accurately and reproducibly, providing a strong foundation for downstream analysis.
Contact us for expert support.